Arin
Az arinek, más néven dehidroarének vagy benzinek[m 1] aromás gyűrűből származtatható rendkívül reakcióképes részecskék, melyek két orto-helyzetű szubsztituensnek a gyűrűből történő eltávolításával keletkeznek.[1][2] Az arinek leginkább feszült hármas kötésükkel jellemezhetőek, de némi kettős gyök karakterrel is rendelkeznek. Az arin kifejezés leginkább az orto-arinekre (1,2-didehidrobenzol) használatos, azonban 1,3- és 1,4-didehidrobenzol köztitermékeket is leírtak már.
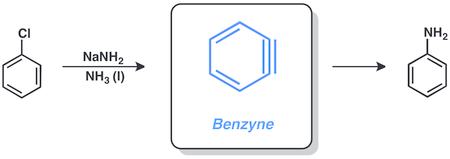
Felfedezését követően gyorsan kifejlesztették azokat a módszereket, melyekkel ezt a rendkívül reakcióképes köztiterméket a szintetikus szerves kémia hasznosítani tudta. Számos természetes vegyületet állítottak elő arin köztiterméken keresztül.[3]
Kötésrendszer
szerkesztésA dehidrobenzol (benzin) alkinként történő megjelenítése a legáltalánosabb, azonban a kumulén és kettős gyök szerkezetek is jelentős mezomer szerkezetek.[4]

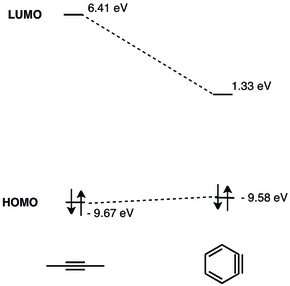
Az orto-dehidrobenzol hármas kötésével járó geometriai korlátok miatt csökken a gyűrű síkjában a p-pályák közötti átfedés, ami csökkenti a hármas kötés erősségét.[5] A dehidrobenzol hármas kötésének rezgési frekvenciáját Rasziszhewski 1846 cm −1 hullámszámhoz asszignálta,[6] amely a telítetlen alkinekre jellemző körülbelül 2150 cm−1-es értékhez képest jóval gyengébb hármas kötést jelez. Mindazonáltal a feszült alkin szerkezet jobban leírja az orto-dehidrobenzolt (nagyobb különbség a szingulett-triplett energiaszintek között, illetve alkinszerű reakciókészség), mint a kettős gyök modell.[7] A geometriai korlátok miatt az arinek LUMO energiaszintje jelentősen lecsökken (a 2-butinban 6,41 eV, míg a dehidrobenzolban csak 1,33 eV), míg a HOMO energiaszint a számítások szerint lényegében változatlan marad.[5][8]

Az arinek LUMO pályája jóval alacsonyabb energiájú, mint a feszültség nélküli alkineké, így energetikailag jobban illik a nukleofilek HOMO pályájához. A dehidrobenzol emiatt elektrofil sajátságú és nukleofilekkel reagál.[9] A dehidrobenzol részletes MO-elemzését 1968-ban mutatták be.[10]
Felfedezése
szerkesztésAz arin típusú köztitermék létezésének első jele Stoermer és Kahlert 1902-es munkájából származik: megfigyelték, hogy 3-brómbenzofuránt etanolban bázissal reagáltatva 2-etoxibenzofurán keletkezik. Az észleltek alapján egy arin köztitermék létezését tételezték fel.[11]

Georg Wittig és munkatársai azt feltételezték, hogy a bifenil fluorbenzolból és fenillítiumból ikerionos köztiterméken át keletkezik.[12][13][14] Ezt kísérletileg John D. Roberts igazolta 1953-ban.[15][16][17][18][19]

1953-ban John D. Roberts elvégezte klasszikussá vált 14C nyomjelzős kísérletét, melynek eredménye erősen alátámasztotta a dehidrobenzol létezését.[15] Roberts és tanítványai klórbenzol-1-14C-et reagáltattak kálium-amiddal, és azt vizsgálták, hogy a termékként kapott anilinbe a 14C-nyomjelző hova épült be. Megfigyeléseik szerint ugyanakkora mennyiségben keletkezett a C-1 és C-2 helyzetű 14C-et tartalmazó termék. Ilyen eredményhez szimmetrikus köztitermék szükséges – ezt ma dehidrobenzolként ismerjük.

Nem sokkal Roberték felfedezése után Wittig és Pohmer felfedezte, hogy a dehidrobenzol részt tud venni [4+2] cikloaddíciós reakciókban.[20]

A dehidrobenzol létezését spektroszkópiai vizsgálatok – IR,[21] UV/látható,[22] mikrohullámú[23] és NMR[24][25] – is megerősítették.
A dehidrobenzolt „molekuláris konténerben” vizsgálták.[26][27]
Arinek előállítása
szerkesztésAz arineket – rendkívüli reakciókészségük miatt – in situ kell előállítani. Az arinkémia kezdetén nagyon erélyes reakciókörülményeket kellett alkalmazni – számos módszer erős bázisokat vagy magas hőmérsékletet igényel. Aril-halogenideket erős bázissal kezelve az aromás gyűrűről el lehet távolítani egy protont, így eliminációval dehidrobenzol állítható elő.

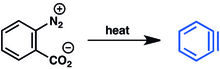
Aréndiazónium-2-karboxilát vegyületek is használhatók kiindulási anyagaként. E módszer fő hátránya a diazóniumvegyületek robbanásra való hajlama.
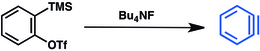
A dehidrobenzol előállítására enyhébb módszereket is kifejlesztettek. Ilyen szintézisre elterjedten használják az aril-triflátokat.[3] A trimetilszililcsoport fluoriddal történő – alább bemutatott – lecserélésével enyhe körülmények között állítható elő dehidrobenzol.
A hexadehidro-Diels–Alder-reakció (HDDA) 1,3-diin és alkin cikloaddíciós reakciója, ezzel szintén elő lehet állítani dehidrobenzolt.[28]

Az arinek reakciói
szerkesztésAz arinek még alacsony hőmérsékleten is rendkívül reakcióképesek. Reakcióik négy fő csoportba sorolhatók: (1) nukleofil addíció, (2) periciklusos reakció, (3) beékelődéses (bond-insertion) reakció és (4) fémkatalizált reakció.
Nukleofil addíció arinekre
szerkesztésBázikus nukleofilekkel kezelve az aril-halogenidek a távozó csoporthoz képest alfa-helyzetben deprotonálódnak, ami összességében egy dehidrohalogénezési reakciónak felel meg. A keletkező dehidrobenzol – jellemzően kezdeti protonálódást követően – addíciós termékekké alakul. A folyamatban a dehidrobenzol köztitermék keletkezése a lassú lépés.[29]
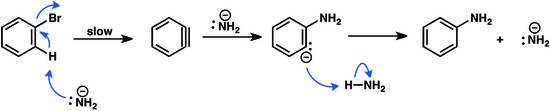
Az „arin kapcsolási” reakciókkal bifenil származékokat lehet előállítani, melyeket a gyógyszeriparban, a mezőgazdaságban és számos fémkatalizált reakcióban ligandumként hasznosítanak.[30]

Szubsztituens hatása
szerkesztésA hármas kötés keletkezésének regiokémiája
szerkesztésHa az (LG) távozó csoport és az (Y) szubsztituens egymáshoz képest orto vagy para helyzetű, akkor csak egyetlen dehidrobenzol köztitermék keletkezhet. Meta helyzet esetén azonban két regiokémiai kimenetel is lehetséges (A és B). Ha Y elektronszívó csoport, akkor HB savasabb, mint HA, így a B regioizomer keletkezik. Hasonló módon, ha Y elektronküldő csoport, akkor az A regioizomer képződik, mivel ekkor HA a savasabb proton.

A hármas kötésre történő nukleofil addíció regiokémiája
szerkesztésAz (Y) szubsztituenst tartalmazó dehidrobenzolnak két lehetséges regioizomerje van: a hármas kötés vagy a C2 és a C3, vagy a C3 és a C4 atomok között helyezkedhet el. A távozó csoporthoz képest orto helyzetű szubsztituens esetén a hármas kötés a C2 és C3 között fog kialakulni. Para helyzetű Y és távozó csoport esetén a C3 és C4 közötti hármas kötést tartalmazó regioizomer keletkezik. Meta helyzetű szubsztituensek esetén a fent írtaknak megfelelően mindkét regioizomer keletkezhet. Amennyiben a hármas kötés C2 és C3 között található, akkor az elektronszívó szubsztituensek úgy irányítják a nukleofil addíciót, hogy az elektronszívó csoporthoz a lehető legközelebb alakuljon ki a karbanion. Az elektronküldő csoportok szelektivitása a termékképződésre ugyanakkor nagyon kicsi. Azon regioizomer esetében, amelynél a hármas kötés C3 és C4 között helyezkedik el, a szubsztituens hatása a nukleofil addícióra kisebb, gyakran a para és meta helyzetű termékek keveréke keletkezik.[29]

Példák totálszintézisben történő felhasználásra
szerkesztésAz arinekre történő nukleofil addíciót kiterjedten használják a természetes anyagok totálszintézisében. Az arinek nukleofil addíciói igazából az arinkémia legrégebb óta ismert alkalmazásai közé tartoznak.[3] Az arinre történő nukleofil addíciót használták volna a kriptasztolin (1) és a kriptovolin (2) totálszintézisének próbálkozásában is.[31]

A tetraciklusos (+)-liphagal meroterpenoid szintézisében is szerepelt arin köztitermék,[32] a természetes vegyület utolsó gyűrűjét arin ciklizációs reakcióval alakították ki.[3]

Az arinek többkomponensű reakciói hatékony átalakítási módok, melyekkel gyorsan 1,2-diszubsztituált arének képezhetők. Potenciális hasznosságuk ellenére a természetes anyagok szintézisében kevés példa van az arinek többkomponensű reakciójára.[3] A dehydroaltenuene B szintézise során egy négykomponensű arinkapcsolási reakciót használtak.[33]

Arinek periciklusos reakciói
szerkesztés[4+2] cikloaddíció
szerkesztésAz arinek részt vehetnek [4+2] ciklizációs reakcióban. Az alábbi ábrán a dehidrobenzol és furán közötti Diels–Alder-reakció koncentráló mechanizmusa látható. Az arinek számos [4+2] cikloaddíciós reakciója ugyanakkor feltehetően lépcsőzetes mechanizmus szerint megy végbe.
Klasszikus példa az 1,2,3,4-tetrafenilnaftalin szintézise.[34] A tetrabrómbenzolból butillítiummal és furánnal történő reakcióval tetrahidroantacén állítható elő.[35][36]

Az arinek [4+2] cikloaddíciós reakciót gyakran használják a természetes vegyületek totálszintézisében. Az eljárás fő korlátja azonban az, hogy feszült szerkezetű dién – például furán vagy ciklopentadién – szükséges hozzá.[3] 2009-ben Buszek és munkatársai herbindol A-t szintetizáltak arin [4+2] cikloaddícióval.[37] A 6,7-indolin ciklopentadiénnel [4+2] cikloaddíciós reakcióba lép, mely egy bonyolult tetraciklusos terméket eredményez.

[2+2] cikloaddíció
szerkesztésAz arinek sokféle alkénnel [2+2] cikloaddíciós reakcióba lépnek. Az arinek elektrofil természete miatt ehhez a reakcióhoz az elektronküldő szubsztituensekkel rendelkező alkének a legmegfelelőbbek.[38]
Az arinek [2+2] kémiáját – a nagyfokú melléktermék-képződés miatt – ritkán használják a természetes anyagok totálszintéziséhez,[3] de több ilyenre is van példa. 1982-ben Stevens és munkatársai olyan taxodionszintézisről számoltak be, melynek során arin és ketén acetál közötti [2+2] cikloaddíciós reakciót hajtottak végre.[39]

Arinek beékelődési reakciói
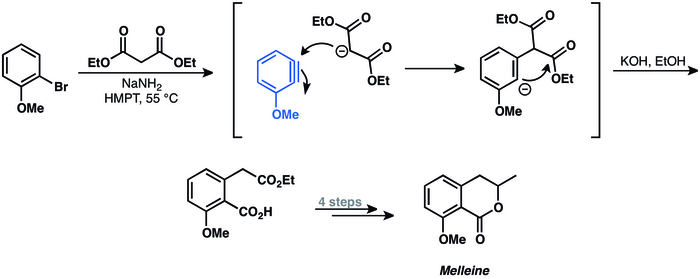
szerkesztésA C−C-kötés beékelődési reakciók az arineket használó módszerek egyik fejlesztési területe. Az első példa egy arin σ-kötése történő beékelődésére a mellein 1973-as szintézise.[40]
Arinek fémkatalizált reakciói
szerkesztésAz arinek fémkatalizált kémiája kevéssé ismert. Mindezidáig csak egyetlen fémkatalizált arin reakciót használnak totálszintézisben.[3] Mori és munkatársai palládium által katalizált arin és diin [2+2+2]-kociklizációs reakciót hajtott végre a taiwanin C totálszintézisére.[41]

További dehidrobenzolok
szerkesztésHa a dehidrobenzol az 1,2-didehidrobenzol, akkor további két izomer is létezhet: 1,3-didehidrobenzol és 1,4-didehidrobenzol.[7] Ezek energiája in silico rendre 106, 122 és 138 kcal/mol (444, 510 és 577 kJ/mol).[42]

Vizsgálták az 1,2-, 1,3- és 1,4-didehidrobenzolok egymásba történő átalakulását.[42][43] Úgy gondolják, hogy a fenil szubsztituált arin prekurzorok pirolízise során (900 °C-on) az 1,2-didehidrobenzol 1,3 izomerré alakul át,[42] ezt az alábbi ábra szemlélteti. A dehidrobenzol izomerizációjához extrém magas hőmérséklet szükséges.

1,4-didehidroarének
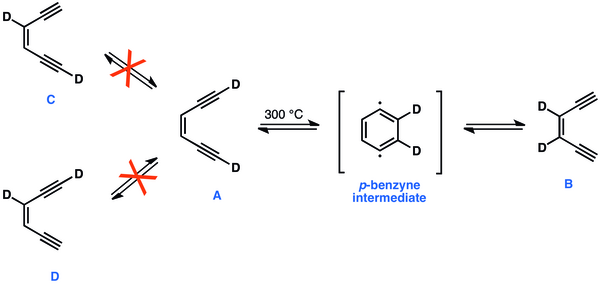
szerkesztésA klasszikus 1,4-didehidrobenzol kísérletekben a 300 °C-ra melegített [1,6-D2]-A könnyen egyensúlyba kerül [3,2-D2]-B-vel, de sem C, sem D nem képződik. A deutériumatomok B formát eredményező egyidejű vándorlása és a tény, hogy sem C, sem D nem keletkezik, csak egy gyűrűs, szimmetrikus köztitermék, az 1,4-di-dehidrobenzol jelenlétével magyarázható.[44]
A Bergman-ciklizáció még fontosabbnak bizonyult az éndiin „citosztatikumok” felfedezésével, ezek a vegyületek ugyanis képesek hasítani a kétszálú DNS-t. Ilyen vegyület például a calicheamicin, melyből a sejtben para-dehidrobenzol származék keletkezik. Az éndiin szerkezetű, potenciálisan tumorellenes/antibiotikus hatású szereknél a biológiai hatás szempontjából kulcsfontosságúnak tartják, hogy a szubsztituált 1,4-didehidrobenzol hidrogénatomot képes elvonni a DNS-ből. Ugyanakkor ezek a molekulák jellemzően nagyon reakcióképesek és nem szelektívek, emiatt eléggé rossz gyógyszerjelöltek. Számítógépes kémiai módszerek segíthetnek az 1,4-didehidrobenzol származékok viselkedésének megjóslásában, ami a jövőben talán hozzájárulhat megfelelő reakciókészségű és nagy szelektivitású gyógyszerjelölt kifejlesztéséhez.[45]
Az 1,4-didehidrobenzolnak két állapotát írták le: egy alacsonyabb energiájú szingulett állapotot és egy triplett állapotot.[46][47] A triplett állapotban két, egymással kölcsönhatásba nem lépő gyökcentrum található, ezért ugyanakkora sebességgel kell a hidrogénatomokat lehasítani, mint a fenilgyöknek. A szingulett állapot ugyanakkor jóval stabilabb, ezért a hidrogén lehasításához ennek a stabilizációs energiának egy részét be kell fektetni, ami kisebb reakciósebességet eredményez. Chen szerint a nagy szingulett-triplett energiakülönbséggel rendelkező 1,4-didehidrobenzol származékok felhasználásával növelni lehetne az éndiin gyógyszerjelöltek szelektivitását.[48] Mindeddig még nem fejlesztettek ki Chen ezen modelljén alapuló gyógyszert.[45]
Megjegyzések
szerkesztés- ↑ A benzin elnevezés a benzol névből és a hármas kötésre utaló -in végződésből származik, nem azonos az üzemanyagként használt benzinnel, lásd Magyar nagylexikon III. (Bah–Bij). Főszerk. Élesztős László, Rostás Sándor. Budapest: Akadémiai. 1994. 635–636. o. ISBN 963-05-6821-7
Hivatkozások
szerkesztés- ↑ Gilchrist T.C.; Rees C.W.; (1969) Carbenes, Nitrenes and Arynes Nelson. London.
- ↑ The Benzyne and Related Intermediates. H. Heaney Chem. Rev., 1962, 62 (2), pp 81–97 doi:10.1021/cr60216a001
- ↑ a b c d e f g h Tadross, P. M.; Stoltz, B. M. A Comprehensive History of Arynes in Natural Product Total SynthesisChem. Rev. 2012, 112, 3550
- ↑ Anslyn, E. V.; Dougherty, D. A.: Modern Physical Organic Chemistry, University Science Books, 2006, p612.
- ↑ a b Gampe, C. M.; Carreira, E. M. Angew Chem. Int. Ed. 2012, 51, 3766
- ↑ Rasziszewski, J. G.; Hess. B. A. J.; Zahradnik, R. J. Am. Chem. Soc. 1992, 114, 52
- ↑ a b Wenk, H. H.; Winkler, M.; Sander, W. Angew. Chem. Int. Ed. 2003, 42, 502
- ↑ Rondan, N. G.; Domelsmith, L. N.; Houk, K.N. Tetrahedron Lett. 1979, 20, 3273
- ↑ Gilchrist, T. L. Supplement C: The Chemistry of Triple Bonded Functional Groups, Part 1. Patai, S.; Rappaport, Z. Eds., John Wiley & Sons, New York, 1983
- ↑ Hoffmann, R.; Imamura, A.;Hehre, W. J. J. Am. Chem. Soc. 1968, 90, 1499
- ↑ „Ueber das 1- und 2-Brom-cumaron”. DOI:10.1002/cber.19020350286. (Hozzáférés: 2013. június 27.)
- ↑ Wittig, G., Pieper, G. and Fuhrmann, G. (1940), Über die Bildung von Diphenyl aus Fluorbenzol und Phenyl-lithium (IV. Mitteil. über Austauschreaktionen mit Phenyl-lithium). Berichte der deutschen chemischen Gesellschaft (A and B Series), 73: 1193–1197. doi:10.1002/cber.19400731113
- ↑ Phenyl-lithium, der Schlüssel zu einer neuen Chemie metallorganischer Verbindungen Georg Wittig Naturwissenschaften, 1942, Volume 30, Numbers 46-47, Pages 696-703 doi:10.1007/BF01489519
- ↑ Wittig, G. (1954), Fortschritte auf dem Gebiet der organischen Aniono-Chemie. Angewandte Chemie, 66: 10–17. doi:10.1002/ange.19540660103
- ↑ a b rearrangement in the reaction of chlorobenzene-1-C14 with potassium amid John D. Roberts, Howard E. Simmons Jr., L. A. Carlsmith, C. Wheaton Vaughan J. Am. Chem. Soc., 1953, 75 (13), pp 3290–3291 doi:10.1021/ja01109a523
- ↑ The Mechanism of Aminations of Halobenzenes John D. Roberts, Dorothy A. Semenow, Howard E. Simmons Jr., L. A. Carlsmith J. Am. Chem. Soc., 1956, 78 (3), pp 601–611 doi:10.1021/ja01584a024
- ↑ Orientation in Aminations of Substituted Halobenzenes John D. Roberts, C. Wheaton Vaughan, L. A. Carlsmith, Dorothy A. Semenow J. Am. Chem. Soc., 1956, 78 (3), pp 611–614 doi:10.1021/ja01584a025
- ↑ Modern Arylation Methods. Edited by Lutz Ackermann 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim ISBN 978-3-527-31937-4
- ↑ The Benzyne and Related Intermediates. H. Heaney Chem. Rev., 1962, 62 (2), pp 81–97 doi:10.1021/cr60216a001
- ↑ Wittig, G.; Pohmer, L. Angew. Chem. 1955, 67(13), 348.
- ↑ Radziszewski, J. G.; Hess, Jr. B. A.; Zahradnik, R. J. Am. Chem. Soc. 1992, 114, 52.
- ↑ Wenthold, P. G.; Squires, R. R.; Lineberger, W. C. J. Am. Chem. Soc. 1998, 120, 5279
- ↑ Kukolich, S. G.; Tanjaroon,C.; McCarthy, M. C.; Thaddeus, P. J. Chem. Phys. 2003, 119, 4353
- ↑ Orendt, A. M.; Facelli, J. C.; Radziszewski, J. G.; Horton, W. J.; Grant, D. M.; Michl, J. J. Am. Chem. Soc. 1996, 118, 846
- ↑ Warmuth,R. Angew. Chem., Int. Ed. Engl. 1997, 36, 1347
- ↑ Warmuth, R. Angew. Chem. Int. Ed. Engl. 1997, 36, 1347
- ↑ Warmuth, R.; Yoon, Acc. Chem. Res. 2001, 34, 96
- ↑ Hoye, T. R.; Baire, B.; Niu, D.; Willoughby, P. H.; Woods, B. P. Nature, 2012, 490, 208
- ↑ a b Anslyn, E. V.; Dougherty, D. A. Modern Physical Organic Chemistry. University Science Books, 2006.
- ↑ Diemer, V.; Begaut, M.; Leroux, F. R.; Colobert, F. Eur. J. Org. Chem. 2011, 341
- ↑ Kametani, T.; Ogasawara, K. J. J. Chem. Soc., C 1967, 2208
- ↑ Day, J. J.; McFadden, R. M.; Virgil, S. C.; Kolding, H.; Alleva, J. L.; Stoltz, B. M. Angew. Chem. Int. Ed. 2011, 50, 6814.
- ↑ Soorukram, D.; Qu, T.; Barrett, A. G. M. Org. Lett. 2008, 10, 3833
- ↑ Organic Syntheses, Coll. Vol. 5, p.1037 (1973); Vol. 46, p.107 (1966). Link
- ↑ Organic Syntheses, Coll. Vol. 10, p.678; Vol. 75, p.201 Article
- ↑ A szin és anti sztereoizomerek keveréke metanolbeli eltérő oldhatóságuk alapján választható el.
- ↑ Buszek, K. R.; Brown, N.; Kuo, D. Org. Lett. 2009, 11, 201
- ↑ Pellissier, H.; Santelli, M. Tetrahedron, 2003, 59, 701
- ↑ Stevens, R. V.; Bisacchi, G. S> J. Org. Chem. 1982, 47, 2396
- ↑ Guyot, M.; Molho, D. Tetrahedron Lett. 1973, 14, 3433
- ↑ Sato, Y.; Tamura,T.; Mori, M. Angew. Chem., Int. Ed. 2004, 43, 2436
- ↑ a b c A m-Benzyne to o-Benzyne Conversion Through a 1,2-Shift of a Phenyl Group. Blake, M. E.; Bartlett, K. L.; Jones, M. Jr. J. Am. Chem. Soc. 2003, 125, 6485. doi:10.1021/ja0213672
- ↑ A p-Benzyne to m-Benzyne Conversion Through a 1,2-Shift of a Phenyl Group. Completion of the Benzyne Cascade, Polishchuk, A. L.; Bartlett, K. L.; Friedman, L. A.; Jones, M. Jr. J. Phys. Org. Chem. 2004, Volume 17, Issue 9 , Pages 798 - 806. doi:10.1002/poc.797
- ↑ Jones, R. R,; Bergman, R. G. J. Am. Chem. Soc. 1972, 94, 660
- ↑ a b Bachrach, S. M. Computational Organic Chemistry. John Wiley & Sons, Inc. 2007
- ↑ Clauberg, H.; Minsek, D. W.; Chen, P. J. Am. Chem. Soc. 1992, 114, 99.
- ↑ Blush, J. A.; Clauberg, H.; Kohn, D. W.; Minsek, D. W.; Zhang, X.; Chen, P. Acc. Chem. Res. 1992, 25, 385
- ↑ Chen, P. Angew. Chem. Int. Ed. Engl. 1996, 35, 1478.
Fordítás
szerkesztésEz a szócikk részben vagy egészben az Aryne című angol Wikipédia-szócikk ezen változatának fordításán alapul. Az eredeti cikk szerkesztőit annak laptörténete sorolja fel. Ez a jelzés csupán a megfogalmazás eredetét és a szerzői jogokat jelzi, nem szolgál a cikkben szereplő információk forrásmegjelöléseként.